 |
 |
 |
|
||||||||||||||||||||||||
 | ||||||||||||||||||||||||
 | ||||||||||||||||||||||||
 | ||||||||||||||||||||||||
Theory of Confocal Microscopy
Fluorophores for Confocal Microscopy
Biological laser scanning confocal microscopy relies heavily on fluorescence as an imaging mode, primarily due to the high degree of sensitivity afforded by the technique coupled with the ability to specifically target structural components and dynamic processes in chemically fixed as well as living cells and tissues. Many fluorescent probes are constructed around synthetic aromatic organic chemicals designed to bind with a biological macromolecule (for example, a protein or nucleic acid) or to localize within a specific structural region, such as the cytoskeleton, mitochondria, Golgi apparatus, endoplasmic reticulum, and nucleus. Other probes are employed to monitor dynamic processes and localized environmental variables, including concentrations of inorganic metallic ions, pH, reactive oxygen species, and membrane potential. Fluorescent dyes are also useful in monitoring cellular integrity (live versus dead and apoptosis), endocytosis, exocytosis, membrane fluidity, protein trafficking, signal transduction, and enzymatic activity. In addition, fluorescent probes have been widely applied to genetic mapping and chromosome analysis in the field of molecular genetics.
The history of synthetic fluorescent probes dates back over a century to the late 1800s when many of the cornerstone dyes for modern histology were developed. Among these were pararosaniline, methyl violet, malachite green, safranin O, methylene blue, and numerous azo (nitrogen) dyes, such as Bismarck brown. Although these dyes were highly colored and capable of absorbing selected bands of visible light, most were only weakly fluorescent and would not be useful for the fluorescence microscopes that would be developed several decades later. However, several synthetic dye classes synthesized during this period, based on the xanthene and acridine heterocyclic ring systems, proved to be highly fluorescent and provided a foundation for the development of modern synthetic fluorescent probes. Most notable among these early fluorescent dyes were the substituted xanthenes, fluorescein and rhodamine B, and the biaminated acridine derivative, acridine orange.
Fluorochromes were introduced to fluorescence microscopy in the early twentieth century as vital stains for bacteria, protozoa, and trypanosomes, but did not see widespread use until the 1920s when fluorescence microscopy was first used to study dye binding in fixed tissues and living cells. However, it wasn't until the early 1940s that Albert Coons developed a technique for labeling antibodies with fluorescent dyes, thus giving birth to the field of immunofluorescence. Over the past 60 years, advances in immunology and molecular biology have produced a wide spectrum of secondary antibodies and provided insight into the molecular design of fluorescent probes targeted at specific regions within macromolecular complexes.
Fluorescent probe technology and cell biology were dramatically altered by the discovery of the green fluorescent protein (GFP) from jellyfish and the development of mutant spectral variants, which have opened the door to non-invasive fluorescence multicolor investigations of subcellular protein localization, intermolecular interactions, and trafficking using living cell cultures. More recently, the development of nanometer-sized fluorescent semiconductor quantum dots has provided a new avenue for research in confocal and widefield fluorescence microscopy. Despite the numerous advances made in fluorescent dye synthesis during the past few decades, there is very little solid evidence about molecular design rules for developing new fluorochromes, particularly with regard to matching absorption spectra to available confocal laser excitation wavelengths. As a result, the number of fluorophores that have found widespread use in confocal microscopy is a limited subset of the many thousands that have been discovered.
Important Characteristics of Fluorophores
Fluorophores are catalogued and described according to their absorption and fluorescence properties, including the spectral profiles, wavelengths of maximum absorbance and emission, and the fluorescence intensity of the emitted light. One of the most useful quantitative parameters for characterizing absorption spectra is the molar extinction coefficient (denoted with the Greek symbol e, see Figure 2(a)), which is a direct measure of the ability of a molecule to absorb light. The extinction coefficient is useful for converting units of absorbance into units of molar concentration, and is determined by measuring the absorbance at a reference wavelength (usually the maximum, characteristic of the absorbing species) for a molar concentration in a defined optical path length. The quantum yield of a fluorochrome or fluorophore represents a quantitative measure of fluorescence emission efficiency, and is expressed as the ratio of the number of photons emitted to the number of photons absorbed. In other words, the quantum yield represents the probability that a given excited fluorochrome will produce an emitted (fluorescence) photon. Quantum yields typically range between a value of 0 and 1, and fluorescent molecules commonly employed as probes in microscopy have quantum yields ranging from very low (0.05 or less) to almost unity. In general, a high quantum yield is desirable in most imaging applications. The quantum yield of a given fluorophore varies, sometimes to large extremes, with environmental factors, such as metallic ion concentration, pH, and solvent polarity.
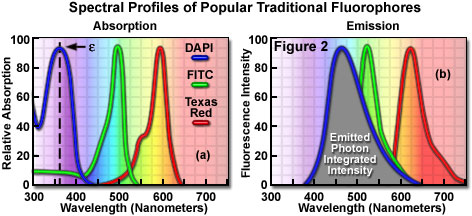
In most cases, the molar extinction coefficient for photon absorption is quantitatively measured and expressed at a specific wavelength, whereas the quantum efficiency is an assessment of the total integrated photon emission over the entire spectral band of the fluorophore (see Figure 2(b)). As opposed to traditional arc-discharge lamps used with the shortest range (10-20 nanometers) bandpass interference filters in widefield fluorescence microscopy, the laser systems used for fluorophore excitation in scanning confocal microscopy restrict excitation to specific laser spectral lines that encompass only a few nanometers. The fluorescence emission spectrum for both techniques, however, is controlled by similar bandpass or longpass filters that can cover tens to hundreds of nanometers. Below saturation levels, fluorescence intensity is proportional to the product of the molar extinction coefficient and the quantum yield of the fluorophore, a relationship that can be utilized to judge the effectiveness of emission as a function of excitation wavelength(s). These parameters display approximately a 20-fold range in variation for the popular fluorophores commonly employed for investigations in confocal microscopy with quantum yields ranging from 0.05 to 1.0, and extinction coefficients ranging from ten thousand to a quarter million (liters per mole). In general, the absorption spectrum of a fluorophore is far less dependent upon environmental conditions than the fluorescence emission characteristics (spectral wavelength profile and quantum yield).
Fluorophores chosen for confocal applications must exhibit a brightness level and signal persistence sufficient for the instrument to obtain image data that does not suffer from excessive photobleaching artifacts and low signal-to-noise ratios. In widefield fluorescence microscopy, excitation illumination levels are easily controlled with neutral density filters, and the intensity can be reduced (coupled with longer emission signal collection periods) to avoid saturation and curtail irreversible loss of fluorescence. Excitation conditions in confocal microscopy are several orders of magnitude more severe, however, and restrictions imposed by characteristics of the fluorophores and efficiency of the microscope optical system become the dominating factor in determining excitation rate and emission collection strategies.
Laser and Arc-Discharge Spectral Lines in Widefield and Confocal Microscopy
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Table 1
Because of the narrow and wavelength-restricted laser spectral lines employed to excite fluorophores in confocal microscopy (see Table 1), fluorescence emission intensity can be seriously restricted due to poor overlap of the excitation wavelengths with the fluorophore absorption band. In addition, the confocal pinhole aperture, which is critical in obtaining thin optical sections at high signal-to-noise ratios, is responsible for a 25 to 50 percent loss of emission intensity, regardless of how much effort has been expended on fine-tuning and alignment of the microscope optical system. Photomultiplier tubes are the most common detectors in confocal microscopy, but suffer from a quantum efficiency that varies as a function of wavelength (especially in the red and infrared regions), further contributing to a wavelength-dependent loss of signal across the emission spectrum. Collectively, the light losses in confocal microscopy can result in a reduction of intensity exceeding 50 times of the level typically observed in widefield fluorescence instruments. It should be clear from the preceding argument that fluorophore selection is one of the most critical aspects of confocal microscopy, and instrumental efficiency must be carefully considered, as well, in order to produce high quality images.
In confocal microscopy, irradiation of the fluorophores with a focused laser beam at high power densities increases the emission intensity up to the point of dye saturation, a condition whose parameters are dictated by the excited state lifetime. In the excited state, fluorophores are unable to absorb another incident photon until they emit a lower-energy photon through the fluorescence process. When the rate of fluorophore excitation exceeds the rate of emission decay, the molecules become saturated and the ground state population decreases. As a result, a majority of the laser energy passes through the specimen undiminished and does not contribute to fluorophore excitation. Balancing fluorophore saturation with laser light intensity levels is, therefore, a critical condition for achieving the optimal signal-to-noise ratio in confocal experiments.
The number of fluorescent probes currently available for confocal microscopy runs in the hundreds, with many dyes having absorption maxima closely associated with common laser spectral lines. An exact match between a particular laser line and the absorption maximum of a specific probe is not always possible, but the excitation efficiency of lines near the maximum is usually sufficient to produce a level of fluorescence emission that can be readily detected. For example, in Figure 3 the absorption spectra of two common probes are illustrated, along with the most efficient laser excitation lines. The green spectrum is the absorption profile of fluorescein isothiocyanate (FITC), which has an absorption maximum of 495 nanometers. Excitation of the FITC fluorophore at 488 nanometers using an argon-ion laser produces an emission efficiency of approximately 87 percent. In contrast, when the 477-nanometer or the 514-nanometer argon-ion laser lines are used to excite FITC, the emission efficiency drops to only 58 or 28 percent, respectively. Clearly, the 488-nanometer argon-ion (or krypton-argon) laser line is the most efficient source for excitation of this fluorophore.
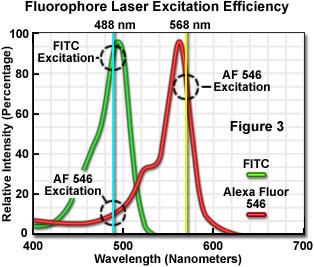
The red spectrum in Figure 3 is the absorption profile of Alexa Fluor 546, a bi-sulfonated alicyclic xanthene (rhodamine) derivative with a maximum extinction coefficient at 556 nanometers, which is designed specifically to display increased quantum efficiency at significantly reduced levels of photobleaching in fluorescence experiments. The most efficient laser excitation spectral line for Alexa Fluor 546 is the yellow 568-nanometer line from the krypton-argon mixed gas ion laser, which produces an emission efficiency of approximately 84 percent. The next closest laser spectral lines, the 543-nanometer line from the green helium-neon laser and the 594-nanometer lines from the yellow helium-neon laser, excite Alexa Fluor 546 with an efficiency of 43 and 4 percent, respectively. Note that the 488-nanometer argon-ion laser spectral line excites Alexa Fluor 546 with approximately 7-percent efficiency, a factor that can be of concern when conducting dual labeling experiments with FITC and Alexa Fluor 546 simultaneously.
Instrumentally, fluorescence emission collection can be optimized by careful selection of objectives, detector aperture dimensions, dichromatic and barrier filters, as well as maintaining the optical train in precise alignment. In most cases, low magnification objectives with a high numerical aperture should be chosen for the most demanding imaging conditions because light collection intensity increases as the fourth power of the numerical aperture, but only decreases as the square of the magnification. However, the most important limitations in light collection efficiency in confocal microscopy arise from restrictions imposed by the physical properties of the fluorophores themselves. As previously discussed, Fluorescent probe development is limited by a lack of knowledge of the specific molecular properties responsible for producing optimum fluorescence characteristics, and the design rules are insufficiently understood to be helpful as a guide to the development of more efficient fluorophores. The current success in development of new fluorescent probes capable of satisfactory performance in confocal microscopy is a testament to the progress made through use of empirical data and assumptions about molecular structure extrapolated from the properties of existing dyes, many of which were first synthesized over a hundred years ago.
Traditional Fluorescent Dyes
The choice of fluorescent probes for confocal microscopy must address the specific capabilities of the instrument to excite and detect fluorescence emission in the wavelength regions made available by the laser systems and detectors. Although the current lasers used in confocal microscopy (see Table 1) produce discrete lines in the ultraviolet, visible, and near-infrared portions of the spectrum, the location of these spectral lines does not always coincide with absorption maxima of popular fluorophores. In fact, it is not necessary for the laser spectral line to correspond exactly with the fluorophore wavelength of maximum absorption, but the intensity of fluorescence emission is regulated by the fluorophore extinction coefficient at the excitation wavelength (as discussed above). The most popular lasers for confocal microscopy are air-cooled argon and krypton-argon ion lasers, the new blue diode lasers, and a variety of helium-neon systems. Collectively, these lasers are capable of providing excitation at ten to twelve specific wavelengths between 400 and 650 nanometers.
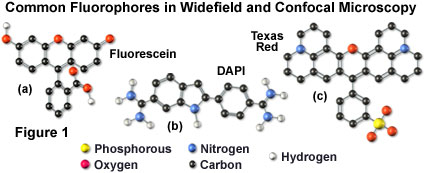
Many of the classical fluorescent probes that have been successfully utilized for many years in widefield fluorescence, including fluorescein isothiocyanate (see Figure 1(a), Figure 2, and Figure 3), Lissamine rhodamine, and Texas red (structure illustrated in Figure 1(c); spectra in Figure 2), are also useful in confocal microscopy. Fluorescein is one of the most popular fluorochromes ever designed, and has enjoyed extensive application in immunofluorescence labeling. This xanthene dye has an absorption maximum at 495 nanometers (see Figure 2(a) and Figure 3), which coincides quite well with the 488 nanometer (blue) spectral line produced by argon-ion and krypton-argon lasers, as well as the 436 and 467 principal lines of the mercury and xenon arc-discharge lamps (respectively). In addition, the quantum yield of fluorescein is very high and a significant amount of information has been gathered on the characteristics of this dye with respect to the physical and chemical properties. On the negative side, the fluorescence emission intensity of fluorescein is heavily influenced by environmental factors (such as pH), and the relatively broad emission spectrum often overlaps with those of other fluorophores in dual and triple labeling experiments.
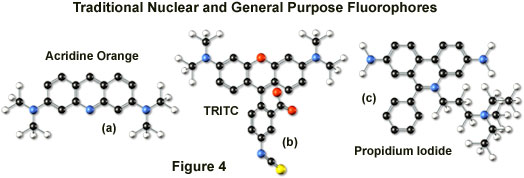
Tetramethyl rhodamine (TMR) and the isothiocyanate derivative (TRITC; Figure 4(b)) are frequently employed in multiple labeling investigations in widefield microscopy due to their efficient excitation by the 546 nanometer spectral line from mercury arc-discharge lamps. The fluorochromes, which have significant emission spectral overlap with fluorescein, can be excited very effectively by the 543 nanometer line from helium-neon lasers, but not by the 514 or 568 nanometer lines from argon-ion and krypton-argon lasers. When using krypton-based laser systems, Lissamine rhodamine is a far better choice in this fluorochrome class due to the absorption maximum at 575 nanometers and its spectral separation from fluorescein. Also, the fluorescence emission intensity of rhodamine derivatives is not as dependent upon strict environmental conditions as that of fluorescein.
Several of the acridine dyes, first isolated in the nineteenth century, are useful as fluorescent probes in confocal microscopy. The most widely utilized, acridine orange, consists of the basic acridine nucleus with dimethylamino substituents located at the 3 and 6 positions of the tri-nuclear ring system (as illustrated in Figure 4(a)). In physiological pH ranges, the molecule is protonated at the heterocyclic nitrogen and exists predominantly as a cationic species in solution. Acridine orange binds strongly to DNA by intercalation of the acridine nucleus between successive base pairs, and exhibits green fluorescence with a maximum wavelength of 530 nanometers. The probe also binds strongly to RNA or single-stranded DNA, but has a longer wavelength fluorescence maximum (approximately 640 nanometers; red) when bound to these macromolecules. In living cells, acridine orange diffuses across the cell membrane (by virtue of the association constant for protonation) and accumulates in the lysosomes and other acidic vesicles. Similar to most acridines and related polynuclear nitrogen heterocycles, acridine orange has a relatively broad absorption spectrum, which enables the probe to be used with several wavelengths from the argon-ion laser.
Another popular traditional probe that is useful in confocal microscopy is the phenanthridine derivative, propidium iodide, first synthesized as an anti-trypanosomal agent along with the closely related ethidium bromide (Figure 4(c)). Propidium iodide binds to DNA in a manner similar to the acridines (via intercalation) to produce orange-red fluorescence centered at 617 nanometers. The positively charged fluorophore also has a high affinity for double-stranded RNA. Propidium has an absorption maximum at 536 nanometers, and can be excited by the 488-nanometer or 514-nanometer spectral lines of an argon-ion (or krypton-argon) laser, or the 543-nanometer line from a green helium-neon laser. The dye is often employed as a counterstain to highlight cell nuclei during double or triple labeling of multiple intracellular structures. Environmental factors can affect the fluorescence spectrum of propidium, especially when the dye is used with mounting media containing glycerol. The structurally similar ethidium bromide, which also binds to DNA by intercalation, produces more background staining and is therefore not as effective as propidium.
DNA and chromatin can also be stained with dyes that bind externally to the double helix. The most popular fluorochromes in this category are 4',6-diamidino-2-phenylindole (DAPI; see Figure 1 and Figure 2) and the bisbenzimide Hoechst dyes that are designated by the numbers 33258, 33342, and 34580. These probes are quite water-soluble and bind externally to AT-rich base pair clusters in the minor groove of double-stranded DNA with a dramatic increase in fluorescence intensity. Both dye classes can be stimulated by the 351-nanometer spectral line of high-power argon-ion lasers or the 354-nanometer line from a helium-cadmium laser. Similar to the acridines and phenanthridines, these fluorescent probes are popular choices as a nuclear counterstain for use in multicolor fluorescent labeling protocols. The vivid blue fluorescence emission produces dramatic contrast when coupled to green, yellow, and red probes in adjacent cellular structures.
Alexa Fluor Dyes
The dramatic advances in modern fluorophore technology are exemplified by the Alexa Fluor dyes introduced by Molecular Probes (Alexa Fluor is a registered trademark of Molecular Probes). These sulfonated rhodamine derivatives exhibit higher quantum yields for more intense fluorescence emission than spectrally similar probes, and have several additional improved features, including enhanced photostability, absorption spectra matched to common laser lines, pH insensitivity, and a high degree of water solubility. In fact, the resistance to photobleaching of Alexa Fluor dyes is so dramatic that even when subjected to irradiation by high-intensity laser sources, fluorescence intensity remains stable for relatively long periods of time in the absence of antifade reagents. This feature enables the water soluble Alexa Fluor probes to be readily utilized for both live-cell and tissue section investigations, as well as in traditional fixed preparations.
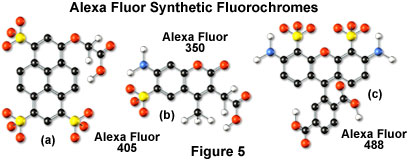
Alexa Fluor dyes are available in a broad range of fluorescence excitation and emission wavelength maxima, ranging from the ultraviolet and deep blue to the near-infrared regions. Alphanumeric names of the individual dyes are associated with the specific excitation laser or arc-discharge lamp spectral lines for which the probes are intended. For example, Alexa Fluor 488 (illustrated in Figure 5(c)) is designed for excitation by the blue 488-nanometer line of the argon or krypton-argon ion lasers, while Alexa Fluor 568 is matched to the 568-nanometer spectral line of the krypton-argon laser. Several of the Alexa Fluor dyes are specifically designed for excitation by either the blue diode laser (405 nanometers; see the Alexa Fluor 405 chemical structure in Figure 5(a)), the orange/yellow helium-neon laser (594 nanometers), or the red helium-neon laser (633 nanometers). Other Alexa Fluor dyes are intended for excitation with traditional mercury arc-discharge lamps in the visible (Alexa Fluor 546) or ultraviolet (Alexa Fluor 350, also useful with high-power argon-ion lasers; Figure 5(b)), and solid-state red diode lasers (Alexa Fluor 680). Because of the large number of available excitation and emission wavelengths in the Alexa Fluor series, multiple labeling experiments can often be conducted exclusively with these dyes.
Alexa Fluor dyes are commercially available as reactive intermediates in the form of maleimides, succinimidyl esters, and hydrazides, as well as prepared cytoskeletal probes (conjugated to phalloidin, G-actin, and rabbit skeletal muscle actin) and conjugates to lectin, dextrin, streptavidin, avidin, biocytin, and a wide variety of secondary antibodies. In the latter forms, the Alexa Fluor fluorophores provide a broad palette of tools for investigations in immunocytochemistry, neuroscience, and cellular biology. The family of probes has also been extended into a series of dyes having overlapping fluorescence emission maxima targeted at sophisticated confocal microscopy detection systems with spectral imaging and linear unmixing capabilities. For example, Alexa Fluor 488, Alexa Fluor 500, and Alexa Fluor 514 are visually similar in color with bright green fluorescence, but have spectrally distinct emission profiles. In addition, the three fluorochromes can be excited with the 488 or 514-nanometer spectral line from an argon-ion laser and are easily detected with traditional fluorescein filter combinations. In multispectral (x-y-l; referred to as a lambda stack) confocal imaging experiments, optical separation software can be employed to differentiate between the similar signals. The overlapping emission spectra of Alexa Fluor 488, 500, and 514 are segregated into separate channels and differentiated using pseudocolor techniques when the three fluorophores are simultaneously combined in a triple label investigation.
Cyanine Dyes
The popular family of cyanine dyes, Cy2, Cy3, Cy5, Cy7, and their derivatives, are based on the partially saturated indole nitrogen heterocyclic nucleus with two aromatic units being connected via a polyalkene bridge of varying carbon number. These probes exhibit fluorescence excitation and emission profiles that are similar to many of the traditional dyes, such as fluorescein and tetramethylrhodamine, but with enhanced water solubility, photostability, and higher quantum yields. Most of the cyanine dyes are more environmentally stable than their traditional counterparts, rendering their fluorescence emission intensity less sensitive to pH and organic mounting media. In a manner similar to the Alexa Fluors, the excitation wavelengths of the Cy series of synthetic dyes are tuned specifically for use with common laser and arc-discharge sources, and the fluorescence emission can be detected with traditional filter combinations.
Marketed by a number of distributors, the cyanine dyes are readily available as reactive dyes or fluorophores coupled to a wide variety of secondary antibodies, dextrin, streptavidin, and egg-white avidin. The cyanine dyes generally have broader absorption spectral regions than members of the Alexa Fluor family, making them somewhat more versatile in the choice of laser excitation sources for confocal microscopy. For example, using the 547-nanometer spectral line from an argon-ion laser, Cy2 is about twice as efficient in fluorescence emission as Alexa Fluor 488. In an analogous manner, the 514-nanometer argon-ion laser line excites Cy3 with a much higher efficiency than Alexa Fluor 546, a spectrally similar probe. Emission profiles of the cyanine dyes are comparable in spectral width to the Alexa Fluor series.
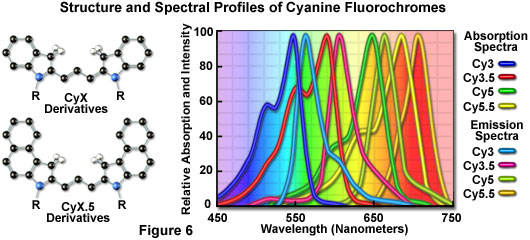
Included in the cyanine dye series are the long-wavelength Cy5 derivatives, which are excited in the red region (650 nanometers) and emit in the far-red (680 nanometers) wavelengths. The Cy5 fluorophore is very efficiently excited by the 647-nanometer spectral line of the krypton-argon laser, the 633-nanometer line of the red helium-neon laser, or the 650-nanometer line of the red diode laser, providing versatility in laser choice. Because the emission spectral profile is significantly removed from traditional fluorophores excited by ultraviolet and blue illumination, Cy5 is often utilized as a third fluorophore in triple labeling experiments. However, similar to other probes with fluorescence emission in the far-red spectral region, Cy5 is not visible to the human eye and can only be detected electronically (using a specialized CCD camera system or photomultiplier). Therefore, the probe is seldom used in conventional widefield fluorescence experiments.
Fluorescent Environmental Probes
Fluorophores designed to probe the internal environment of living cells have been widely examined by a number of investigators, and many hundreds have been developed to monitor such effects as localized concentrations of alkali and alkaline earth metals, heavy metals (employed biochemically as enzyme cofactors), inorganic ions, thiols and sulfides, nitrite, as well as pH, solvent polarity, and membrane potential. Originally, the experiments in this arena were focused on changes in the wavelength and/or intensity of absorption and emission spectra exhibited by fluorophores upon binding calcium ions in order to measure intracellular flux densities. These probes bind to the target ion with a high degree of specificity to produce the measured response and are often referred to as spectrally sensitive indicators. Ionic concentration changes are determined by the application of optical ratio signal analysis to monitor the association equilibrium between the ion and its host. The concentration values derived from this technique are largely independent of instrumental variations and probe concentration fluctuations due to photobleaching, loading parameters, and cell retention. In the past few years, a number of new agents have been developed that bind specific ions or respond with measurable features to other environmental conditions.
Calcium is a metabolically important ion that plays a vital role in cellular response to many forms of external stimuli. Because transient fluctuations in calcium ion concentration are typically involved when cells undergo a response, fluorophores must be designed to measure not only localized concentrations within segregated compartments, but should also produce quantitative changes when flux density waves progress throughout the entire cytoplasm. Many of the synthetic molecules designed to measure calcium levels are based on the non-fluorescent chelation agents EGTA and BAPTA, which have been used for years to sequester calcium ions in buffer solutions. Two of the most common calcium probes are the ratiometric indicators fura-2 and indo-1, but these fluorophores are not particularly useful in confocal microscopy. The dyes are excited by ultraviolet light and exhibit a shift in the excitation or emission spectrum with the formation of isosbestic points when binding calcium. However, the optical aberrations associated with ultraviolet imaging, limited specimen penetration depths, and the expense of ultraviolet lasers have limited the utility of these probes in confocal microscopy.
Fluorophores that respond in the visible range to calcium ion fluxes are, unfortunately, not ratiometric indicators and do not exhibit a wavelength shift (typical of fura-2 and indo-1) upon binding, although they do undergo an increase or decrease in fluorescence intensity. The best example is fluo-3, a complex xanthene derivative, which undergoes a dramatic increase in fluorescence emission at 525 nanometers (green) when excited by the 488-nanometer spectral line of an argon-ion or krypton-argon laser. Because isosbestic points are not present to assure the absence of concentration fluctuations, it is impossible to determine whether spectral changes are due to complex formation or a variation in concentration with fluo-3 and similar fluorophores.

To overcome the problems associated with using visible light probes lacking wavelength shifts (and isosbestic points), several of these dyes are often utilized in combination for calcium measurements in confocal microscopy. Fura red, a multi-nuclear imidazole and benzofuran heterocycle, exhibits a decrease in fluorescence at 650 nanometers when binding calcium. A ratiometric response to calcium ion fluxes can be obtained when a mixture of fluo-3 and fura red is excited at 488 nanometers and fluorescence is measured at the emission maxima (525 and 650 nanometers, respectively) of the two probes. Because the emission intensity of fluo-3 increases monotonically while that of fura red simultaneously decreases, an isosbestic point is obtained when the dye concentrations are constant within the localized area being investigated (as illustrated in Figure 7). Another benefit of using these probes together is the ability to measure fluorescence intensity fluctuations with a standard FITC/Texas red interference filter combination.
Quantitative measurements of ions other than calcium, such as magnesium, sodium, potassium and zinc, are conducted in an analogous manner using similar fluorophores. One of the most popular probes for magnesium, mag-fura-2 (structurally similar to fura red), is also excited in the ultraviolet range and presents the same problems in confocal microscopy as fura-2 and indo-1. Fluorophores excited in the visible light region are becoming available for the analysis of many monovalent and divalent cations that exist at varying concentrations in the cellular matrix. Several synthetic organic probes have also been developed for monitoring the concentration of simple and complex anions.
Important fluorescence monitors for intracellular pH include a pyrene derivative known as HPTS or pyranine, the fluorescein derivative, BCECF, and another substituted xanthene termed carboxy SNARF-1. Because many common fluorophores are sensitive to pH in the surrounding medium, changes in fluorescence intensity that are often attributed to biological interactions may actually occur as a result of protonation. In the physiological pH range (pH 6.8 to 7.4), the probes mentioned above are useful for dual-wavelength ratiometric measurements and differ only in dye loading parameters. Simultaneous measurements of calcium ion concentration and pH can often be accomplished by combining a pH indicator, such as SNARF-1, with a calcium ion indicator (for example, fura-2). Other probes have been developed for pH measurements in subcellular compartments, such as the lysosomes, as described below.
Organelle Probes
Fluorophores targeted at specific intracellular organelles, such as the mitochondria, lysosomes, Golgi apparatus, and endoplasmic reticulum, are useful for monitoring a variety of biological processes in living cells using confocal microscopy. In general, organelle probes consist of a fluorochrome nucleus attached to a target-specific moiety that assists in localizing the fluorophore through covalent, electrostatic, hydrophobic or similar types of bonds. Many of the fluorescent probes designed for selecting organelles are able to permeate or sequester within the cell membrane (and therefore, are useful in living cells), while others must be installed using monoclonal antibodies with traditional immunocytochemistry techniques. In living cells, organelle probes are useful for investigating transport, respiration, mitosis, apoptosis, protein degradation, acidic compartments, and membrane phenomena. Cell impermeant fluorophore applications include nuclear functions, cytoskeletal structure, organelle detection, and probes for membrane integrity. In many cases, living cells that have been labeled with permeant probes can subsequently be fixed and counterstained with additional fluorophores in multicolor labeling experiments.
Mitochondrial probes are among the most useful fluorophores for investigating cellular respiration and are often employed along with other dyes in multiple labeling investigations. The traditional probes, rhodamine 123 and tetramethylrosamine, are rapidly lost when cells are fixed and have largely been supplanted by newer, more specific, fluorophores developed by Molecular Probes. These include the popular MitoTracker (see Figure 8(b)) and MitoFluor series of structurally diverse xanthene, benzoxazole, indole, and benzimidazole heterocycles that are available in a variety of excitation and emission spectral profiles. The mechanism of action varies for each of the probes in this series, ranging from covalent attachment to oxidation within respiring mitochondrial membranes.
MitoTracker dyes are retained quite well after cell fixation in formaldehyde and can often withstand lipophilic permeabilizing agents. In contrast, the MitoFluor probes are designed specifically for actively respiring cells and are not suitable for fixation and counterstaining procedures. Another popular mitochondrial probe, entitled JC-1, is useful as an indicator of membrane potential and in multiple staining experiments with fixed cells. This carbocyanine dye exhibits green fluorescence at low concentrations, but can undergo intramolecular association within active mitochondria to produce a shift in emission to longer (red) wavelengths. The change in emission wavelength is useful in determining the ratio of active to non-active mitochondria in living cells.
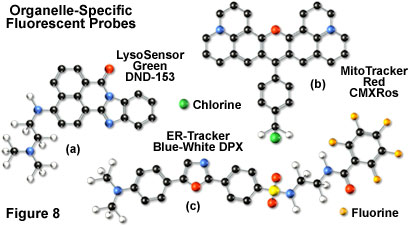
In general, weakly basic amines that are able to pass through membranes are the ideal candidates for investigating biosynthesis and pathogenesis in lysosomes. Traditional lysosomal probes include the non-specific phenazine and acridine derivatives neutral red and acridine orange, which are accumulated in the acidic vesicles upon being protonated. Fluorescently labeled latex beads and macromolecules, such as dextran, can also be accumulated in lysosomes by endocytosis for a variety of experiments. However, the most useful tools for investigating lysosomal properties with confocal microscopy are the LysoTracker and LysoSensor dyes developed by Molecular Probes. These structurally diverse agents contain heterocyclic and aliphatic nitrogen moieties that modulate transport of the dyes into the lysosomes of living cells for both short-term and long-term studies. The LysoTracker probes, which are available in a variety of excitation and emission wavelengths, have high selectivity for acidic organelles and are capable of labeling cells at nanomolar concentrations. Several of the dyes are retained quite well after fixing and permeabilization of cells. In contrast, the LysoSensor fluorophores (Figure 8(a)) are designed for studying dynamic aspects of lysosome function in living cells. Fluorescence intensity dramatically increases in the LysoSensor series upon protonation, making these dyes useful as pH indicators. A variety of Golgi-specific monoclonal antibodies have also been developed for use in immunocytochemistry assays.
Proteins and lipids are sorted and processed in the Golgi apparatus, which is typically stained with fluorescent derivatives of ceramides and sphingolipids. These agents are highly lipophilic, and are therefore useful as markers for the study of lipid transport and metabolism in live cells. Several of the most useful fluorophores for Golgi apparatus contain the complex heterocyclic BODIPY nucleus developed by Molecular Probes. When coupled to sphingolipids, the BODIPY fluorophore is highly selective and exhibits a tolerance for photobleaching that is far superior to many other dyes. In addition, the emission spectrum is dependent upon concentration (shifting from green to red at higher concentrations), making the probes useful for locating and identifying intracellular structures that accumulate large quantities of lipids. During live-cell experiments, fluorescent lipid probes can undergo metabolism to derivatives that may bind to other subcellular features, a factor that can often complicate the analysis of experimental data.
The most popular traditional probes for endoplasmic reticulum fluorescence analysis are the carbocyanine and xanthene dyes, DiOC(6) and several rhodamine derivatives, respectively. These dyes must be used with caution, however, because they can also accumulate in the mitochondria, Golgi apparatus, and other intracellular lipophilic regions. Newer, more photostable, probes have been developed for selective staining of the endoplasmic reticulum by several manufacturers. In particular, oxazole members of the Dapoxyl family produced by Molecular Probes (see Figure 8(c)) are excellent agents for selective labeling of the endoplasmic reticulum in living cells, either alone or in combination with other dyes. These probes are retained after fixation with formaldehyde, but can be lost with permeabilizing detergents. Another useful probe is Brefeldin A, a stereochemically complex fungal metabolite that serves as an inhibitor of protein trafficking out of the endoplasmic reticulum. Finally, similar to other organelles, monoclonal antibodies have been developed that target the endoplasmic reticulum in fixed cells for immunocytochemistry investigations.
Quantum Dots
Nanometer-sized crystals of purified semiconductors known as quantum dots are emerging as a potentially useful fluorescent labeling agent for living and fixed cells in both traditional widefield and laser scanning confocal fluorescence microscopy. Recently introduced techniques enable the purified tiny semiconductor crystals to be coated with a hydrophilic polymer shell and conjugated to antibodies or other biologically active peptides and carbohydrates for application in many of the classical immunocytochemistry protocols. These probes have significant benefits over organic dyes and fluorescent proteins, including long-term photostability, high fluorescence intensity levels, and multiple colors with single-wavelength excitation for all emission profiles.
Quantum dots produce illumination in a manner similar to the well-known semiconductor light emitting diodes, but are activated by absorption of a photon rather than an electrical stimulus. The absorbed photon creates an electron-hole pair that quickly recombines with the concurrent emission of a photon having lower energy. The most useful semiconductor discovered thus far for producing biological quantum dots is cadmium selenide (CdSe), a material in which the energy of the emitted photons is a function of the physical size of the nanocrystal particles. Thus, quantum dots having sizes that differ only by tenths of a nanometer emit different wavelengths of light, with the smaller sizes emitting shorter wavelengths, and vice versa.
Unlike typical organic fluorophores or fluorescent proteins, which display highly defined spectral profiles, quantum dots have an absorption spectrum that increases steadily with decreasing wavelength. Also in contrast, the fluorescence emission intensity is confined to a symmetrical peak with a maximum wavelength that is dependent on the dot size, but independent of the excitation wavelength. As a result, the same emission profile is observed regardless of whether the quantum dot is excited at 300, 400, 500, or 600 nanometers, but the fluorescence intensity increases dramatically at shorter excitation wavelengths. For example, the extinction coefficient for a typical quantum dot conjugate that emits in the orange region (605 nanometers) is approximately 5-fold higher when the semiconductor is excited at 400 versus 600 nanometers. The full width at half maximum value for a typical quantum dot conjugate is about 30 nanometers, and the spectral profile is not skewed towards the longer wavelengths (having higher intensity "tails"), such is the case with most organic fluorochromes. The narrow emission profile enables several quantum dot conjugates to be simultaneously observed with a minimal level of bleed-through.
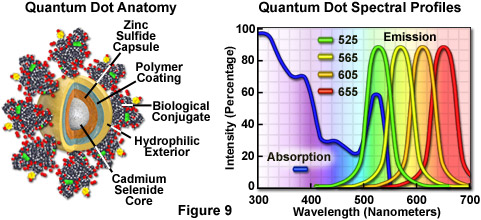
For biological applications, a relatively uniform population of cadmium selenide crystals are covered with a surrounding semiconductor shell composed of zinc sulfide to improve the optical properties. Next, the core material is coated with a polymeric film and other ligands to decrease hydrophobicity and to improve the attachment efficiency of conjugated macromolecules. The final product is a biologically active particle that ranges in size from 10 to 15 nanometers, somewhere in the vicinity of a large protein. Quantum dot conjugates are solubilized as a colloidal suspension in common biological buffers and may be incorporated into existing labeling protocols in place of classical staining reagents (such as organic fluorochrome-labeled secondary antibodies).
In confocal microscopy, quantum dots are excited with varying degrees of efficiency by most of the spectral lines produced by the common laser systems, including the argon-ion, helium-cadmium, krypton-argon, and the green helium-neon. Particularly effective at exciting quantum dots in the ultraviolet and violet regions are the new blue diode and diode-pumped solid-state lasers that have prominent spectral lines at 442 nanometers and below. The 405-nanometer blue diode laser is an economical excitation source that is very effective for use with quantum dots due to their high extinction coefficient at this wavelength. Another advantage of using these fluorophores in confocal microscopy is the ability to stimulate multiple quantum dot sizes (and spectral colors) in the same specimen with a single excitation wavelength, making these probes excellent candidates for multiple labeling experiments.
The exceptional photostability of quantum dot conjugates is of great advantage in confocal microscopy when optical sections are being collected. Unlike the case of organic fluorophores, labeled structures situated away from the focal plane do not suffer from excessive photobleaching during repeated raster scanning of the specimen and yield more accurate three-dimensional volume models. In widefield fluorescence microscopy, quantum dot conjugates are available for use with conventional dye-optimized filter combinations that are standard equipment on many microscopes. Excitation can be further enhanced by substituting a shortpass filter for the bandpass filter that accompanies most filter sets, thus optimizing the amount of lamp energy that can be utilized to excite the quantum dots. Several of the custom fluorescence filter manufacturers offer combinations specifically designed to be used with quantum dot conjugates.
Fluorescent Proteins
Over the past few years, the discovery and development of naturally occurring fluorescent proteins and mutated derivatives have rapidly advanced to center stage in the investigation of a wide spectrum of intracellular processes in living organisms. These biological probes have provided scientists with the ability to visualize, monitor, and track individual molecules with high spatial and temporal resolution in both steady-state and kinetic experiments. A variety of marine organisms have been the source of more than 25 fluorescent proteins and their analogs, which arm the investigator with a balanced palette of non-invasive biological probes for single, dual, and multispectral fluorescence analysis. Among the advantages of fluorescent proteins over the traditional organic and new semiconductor probes described above is their response to a wider variety of biological events and signals. Coupled with the ability to specifically target fluorescent probes in subcellular compartments, the extremely low or absent photodynamic toxicity, and the widespread compatibility with tissues and intact organisms, these biological macromolecules offer an exciting new frontier in live-cell imaging.
The first member of this series to be discovered, green fluorescent protein (GFP), was isolated from the North Atlantic jellyfish, Aequorea victoria, and found to exhibit a high degree of fluorescence without the aid of additional substrates or coenzymes. In native green fluorescent protein, the fluorescent moiety is a tripeptide derivative of serine, tyrosine, and glycine that requires molecular oxygen for activation, but no additional cofactors or enzymes. Subsequent investigations revealed that the GFP gene could be expressed in other organisms, including mammals, to yield fully functional analogs that display no adverse biological effects. In fact, fluorescent proteins can be fused to virtually any protein in living cells using recombinant complementary DNA cloning technology, and the resulting fusion protein gene product expressed in cell lines adapted to standard tissue culture methodology. Lack of a need for cell-specific activation cofactors renders the fluorescent proteins much more useful as generalized probes than other biological macromolecules, such as the phycobiliproteins, which require insertion of accessory pigments in order to produce fluorescence.
Mutagenesis experiments with green fluorescent protein have produced a large number of variants with improved folding and expression characteristics, which have eliminated wild-type dimerization artifacts and fine-tuned the absorption and fluorescence properties. One of the earliest variants, known as enhanced green fluorescence protein (EGFP), contains codon substitutions (commonly referred to as the S65T mutation) that alleviates the temperature sensitivity and increases the efficiency of GFP expression in mammalian cells. Proteins fused with EGFP can be observed at low light intensities for long time periods with minimal photobleaching. Enhanced green fluorescent protein fusion products are optimally excited by the 488-nanometer spectral line from argon and krypton-argon ion lasers in confocal microscopy. This provides an excellent biological probe and instrument combination for examining intracellular protein pathways along with the structural dynamics of organelles and the cytoskeleton.
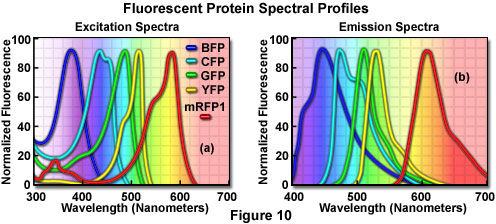
Additional mutation studies have uncovered GFP variants that exhibit a variety of absorption and emission characteristics across the entire visible spectral region, which have enabled researchers to develop probe combinations for simultaneous observation of two or more distinct fluorescent proteins in a single organism (see the spectral profiles in Figure 10). Early investigations yielded the blue fluorescent protein (BFP) and cyan fluorescent protein (CFP) mutants from simple amino acid substitutions that shifted the absorption and emission spectral profiles of wild-type GFP to lower wavelength regions. Used in combination with GFP, these derivatives are useful in resonance energy transfer (FRET) experiments and other investigations that rely on multicolor fluorescence imaging. Blue fluorescent protein can be efficiently excited with the 354-nanometer line from a high-power argon laser, while the more useful cyan derivative is excited by a number of violet and blue laser lines, including the 405-nanometer blue diode, the 442-nanometer helium-cadmium spectral line, and the 457-nanometer line from the standard argon-ion laser.
Another popular fluorescent protein derivative, the yellow fluorescent protein (YFP), was designed on the basis of the GFP crystalline structural analysis to red-shift the absorption and emission spectra. Yellow fluorescent protein is optimally excited by the 514-nanometer spectral line of the argon-ion laser, and provides more intense emission than enhanced green fluorescent protein, but is more sensitive to low pH and high halogen ion concentrations. The enhanced yellow fluorescent protein derivative (EYFP) is useful with the 514 argon-ion laser line, but can also be excited with relatively high efficiency by the 488-nanometer line from argon and krypton-argon lasers. Both of these fluorescent protein derivatives have been widely applied to protein-protein FRET investigations in combination with CFP, and in addition, have proven useful in studies involving multiprotein trafficking.
Attempts to shift the absorption and emission spectra of Aequorea victoria fluorescent proteins to wavelengths in the orange and red regions of the spectrum have met with little success. However, fluorescent proteins from other marine species have enabled investigators to extend the available spectral regions to well within the red wavelength range. The DsRed fluorescent protein and its derivatives, originally isolated from the sea anemone Discosoma striata, are currently the most popular analogs for fluorescence analysis in the 575 to 650-nanometer region. Another protein, HcRed from the Heteractis crispa purple anemone, is also a promising candidate for investigations in the longer wavelengths of the visible spectrum. Newly developed photoactivation fluorescent proteins, including photoactivatable green fluorescent protein (PA-GFP), Kaede, and kindling fluorescent protein 1 (KFP1), exhibit dramatic improvements over GFP (up to several thousand-fold) in fluorescence intensity when stimulated by violet laser illumination. These probes should prove useful in fluorescence confocal studies involving selective irradiation of specific target regions and the subsequent kinetic analysis of diffusional mobility and compartmental residency time of fusion proteins.
Quenching and Photobleaching
The consequences of quenching and photobleaching are suffered in practically all forms of fluorescence microscopy, and result in an effective reduction in the levels of emission. These artifacts should be of primary consideration when designing and executing fluorescence investigations. The two phenomena are distinct in that quenching is often reversible whereas photobleaching is not. Quenching arises from a variety of competing processes that induce non-radiative relaxation (without photon emission) of excited state electrons to the ground state, which may be either intramolecular or intermolecular in nature. Because non-radiative transition pathways compete with the fluorescence relaxation, they usually dramatically lower or, in some cases, completely eliminate emission. Most quenching processes act to reduce the excited state lifetime and the quantum yield of the affected fluorophore.
A common example of quenching is observed with the collision of an excited state fluorophore and another (non-fluorescent) molecule in solution, resulting in deactivation of the fluorophore and return to the ground state. In most cases, neither of the molecules is chemically altered in the collisional quenching process. A wide variety of simple elements and compounds behave as collisional quenching agents, including oxygen, halogens, amines, and many electron-deficient organic molecules. Collisional quenching can reveal the presence of localized quencher molecules or moieties, which via diffusion or conformational change, may collide with the fluorophore during the excited state lifetime. The mechanisms for collisional quenching include electron transfer, spin-orbit coupling, and intersystem crossing to the excited triplet state. Other terms that are often utilized interchangeably with collisional quenching are internal conversion and dynamic quenching.
A second type of quenching mechanism, termed static or complex quenching, arises from non-fluorescent complexes formed between the quencher and fluorophore that serve to limit absorption by reducing the population of active, excitable molecules. This effect occurs when the fluorescent species forms a reversible complex with the quencher molecule in the ground state, and does not rely on diffusion or molecular collisions. In static quenching, fluorescence emission is reduced without altering the excited state lifetime. A fluorophore in the excited state can also be quenched by a dipolar resonance energy transfer mechanism when in close proximity with an acceptor molecule to which the excited state energy can be transferred non-radiatively. In some cases, quenching can occur through non-molecular mechanisms, such as attenuation of incident light by an absorbing species (including the chromophore itself).
In contrast to quenching, photobleaching (also termed fading) occurs when a fluorophore permanently loses the ability to fluoresce due to photon-induced chemical damage and covalent modification. Upon transition from an excited singlet state to the excited triplet state, fluorophores may interact with another molecule to produce irreversible covalent modifications. The triplet state is relatively long-lived with respect to the singlet state, thus allowing excited molecules a much longer timeframe to undergo chemical reactions with components in the environment. The average number of excitation and emission cycles that occur for a particular fluorophore before photobleaching is dependent upon the molecular structure and the local environment. Some fluorophores bleach quickly after emitting only a few photons, while others that are more robust can undergo thousands or even millions of cycles before bleaching.
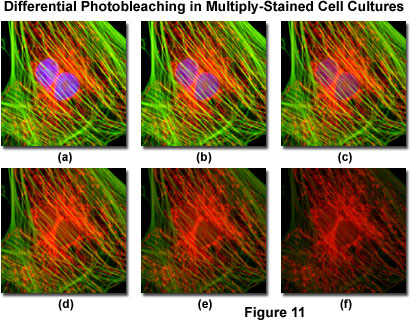
Presented in Figure 11 is a typical example of photobleaching (fading) observed in a series of digital images captured at different time points for a multiply-stained culture of Indian Muntjac deerskin fibroblast cells. The nuclei were stained with DAPI (blue fluorescence), while the mitochondria and actin cytoskeleton were stained with MitoTracker Red (red fluorescence) and an Alexa Fluor phalloidin derivative (Alexa Fluor 488; green fluorescence), respectively. Time points were taken in two-minute intervals using a fluorescence filter combination with bandwidths tuned to excite the three fluorophores simultaneously while also recording the combined emission signals. Note that all three fluorophores have a relatively high intensity in Figure 11(a), but the DAPI (blue) intensity starts to drop rapidly at two minutes and is almost completely gone at six minutes. The mitochondrial and actin stains are more resistant to photobleaching, but the intensity of both drops over the course of the timed sequence (10 minutes).
An important class of photobleaching events are photodynamic, meaning they involve the interaction of the fluorophore with a combination of light and oxygen. Reactions between fluorophores and molecular oxygen permanently destroy fluorescence and yield a free radical singlet oxygen species that can chemically modify other molecules in living cells. The amount of photobleaching due to photodynamic events is a function of the molecular oxygen concentration and the proximal distance between the fluorophore, oxygen molecules, and other cellular components. Photobleaching can be reduced by limiting the exposure time of fluorophores to illumination or by lowering the excitation energy. However, these techniques also reduce the measurable fluorescence signal. In many cases, solutions of fluorophores or cell suspensions can be deoxygenated, but this is not feasible for living cells and tissues. Perhaps the best protection against photobleaching is to limit exposure of the fluorochrome to intense illumination (using neutral density filters) coupled with the judicious use of commercially available antifade reagents that can be added to the mounting solution or cell culture medium.
Under certain circumstances, the photobleaching effect can also be utilized to obtain specific information that would not otherwise be available. For example, in fluorescence recovery after photobleaching (FRAP) experiments, fluorophores within a target region are intentionally bleached with excessive levels of irradiation. As new fluorophore molecules diffuse into the bleached region of the specimen (recovery), the fluorescence emission intensity is monitored to determine the lateral diffusion rates of the target fluorophore. In this manner, the translational mobility of fluorescently labeled molecules can be ascertained within a very small (2 to 5 micrometer) region of a single cell or section of living tissue.
Conclusions
Although the subset of fluorophores that are advantageous in confocal microscopy is rapidly growing, many of the traditional probes that have been useful for years in widefield applications are still of little utility when constrained by fixed-wavelength laser spectral lines. Many of the limitations surrounding the use of fluorophores excited in the ultraviolet region will be eliminated with the introduction of advanced objectives designed to reduce aberration coupled to the gradual introduction of low-cost, high-power diode laser systems with spectral lines in these shorter wavelengths. The 405-nanometer blue diode laser is a rather cheap alternative to more expensive ion and Noble gas-based ultraviolet lasers, and is rapidly becoming available for most confocal microscope systems. Helium-neon lasers with spectral lines in the yellow and orange region have rendered some fluorophores useful that were previously limited to widefield applications. In addition, new diode-pumped solid-state lasers are being introduced with emission wavelengths in the ultraviolet, violet, and blue regions.
Continued advances in fluorophore design, dual-laser scanning, multispectral imaging, and spinning disk applications will also be important in the coming years. The persistent problem of emission crossover due to spectral overlap, which occurs with many synthetic probes and fluorescent proteins in multicolor investigations, benefits significantly from spectral analysis and deconvolution of lambda stacks. Combined, these advances and will dramatically improve the collection and analysis of data obtained from complex fluorescence experiments in live-cell imaging.
Contributing Authors
Jennifer Lippincott-Schwartz - Cell Biology and Metabolism Branch, National Institute of Child Health and Human Development, Building 18T/Room101, 18 Library Drive, National Institutes of Health, Bethesda, Maryland, 20892.
Brian Herman - Department of Cellular and Structural Biology, University of Texas Health Science Center, 7703 Floyd Curl Drive, San Antonio, Texas 78229.
Michael W. Davidson - National High Magnetic Field Laboratory, 1800 East Paul Dirac Dr., The Florida State University, Tallahassee, Florida, 32310.