Confocal Microscopy: Principles, Applications, and Advances
Description :
Dynamic Mode Decomposition (DMD) of FLIP image sequences reveals key intracellular fluorescence dynamics of eGFP-Q145 in CHO cells. (A) Singular values indicate dominant dynamic components. (B) Temporal decays of the two primary modes (rank 2) are shown. (C, D) Real-part spatial maps of mode 0 and mode 1 highlight their distinct intracellular patterns. (E) Reconstructed frames of each mode (every 20th frame) demonstrate the progression of dynamic behaviors, emphasizing DMD’s power to dissect complex fluorescence changes into interpretable spatial-temporal modes.
Confocal microscopy offers significant advantages over conventional widefield optical microscopy, including precise depth-of-field control, reduction of out-of-focus background, and the ability to acquire serial optical sections from thick specimens. By using spatial filtering to exclude light from planes outside the focal point, confocal systems generate sharper, high-contrast images from both fixed and living samples. Its versatility has made confocal microscopy a cornerstone in modern cell and tissue imaging, bridging the gap between traditional fluorescence microscopy and transmission electron microscopy.
Advantages over Widefield Microscopy
In conventional widefield epi-fluorescence microscopy, secondary fluorescence emitted outside the focal plane obscures fine structural details, especially in specimens thicker than 2 μm. Confocal microscopy provides moderate improvements in axial (z-axis) and lateral (x-y plane) resolution while effectively excluding out-of-focus light. This optical sectioning capability dramatically enhances contrast and definition, enabling the visualization of cellular structures that would otherwise be lost in background fluorescence.
For example, thick fluorescently stained human medulla or rabbit muscle fibers appear blurry under widefield imaging but reveal detailed structural features with confocal imaging. Similarly, autofluorescent sunflower pollen grains show clear internal and external morphology when optically sectioned with a confocal microscope.
Historical Development
The concept of confocal microscopy was first introduced by Marvin Minsky in the 1950s to study neural networks in unstained brain tissue. Early adoption was limited by insufficient light sources and computational power. Subsequent developments by M. David Egger, Mojmir Petran, G. Fred Brakenhoff, Colin Sheppard, and others in the 1970s and 1980s refined scanning and imaging methods, eventually leading to commercial instruments in 1987. Advances in lasers, optics, detectors, and digital computing throughout the 1990s fueled a rapid expansion of confocal microscopy applications.
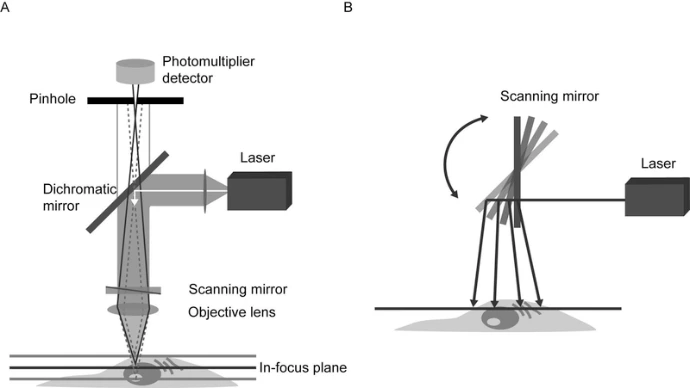
Principles of Confocal Microscopy
A confocal microscope uses laser light focused to a diffraction-limited spot and scanned across the specimen. Secondary fluorescence from the focal plane passes through a pinhole to a detector, while out-of-focus light is mostly blocked. Key components include:
Laser excitation source: Provides coherent light for fluorescence excitation.
Scan head: Raster-scans the laser and collects emitted photons.
Pinhole aperture: Acts as a spatial filter, eliminating out-of-focus fluorescence.
Photodetector: Converts emitted light into an analog signal for digitization.
Computer system: Processes, analyzes, and displays images.
Unlike widefield microscopy, confocal imaging is point-by-point, ensuring precise optical sectioning. Depth (z-axis) resolution typically ranges from 0.5 to 1.5 μm, depending on objective, laser wavelength, and pinhole size.
Modern Confocal Systems
Today’s confocal microscopes integrate multiple lasers, detectors, scanning mirrors, and computers to provide high-resolution, multi-dimensional imaging. Specimens can be scanned either by moving the stage or scanning the laser beam. Single-beam galvanometer scanning and multi-beam Nipkow disk systems are the most common approaches. Multi-beam systems allow real-time imaging with reduced photodamage, while single-beam systems provide high-resolution optical sectioning.
Laser scanning confocal microscopes can generate:
Serial optical sections: Thin slices through thick specimens for 3D reconstruction.
3D volume-rendered images: Visualizing internal structures of cells, tissues, or pollen grains.
Multi-dimensional, multi-color imaging: Tracking dynamic processes in living cells.
Additional features include electronic zoom, flexible magnification, and precise digital image processing, enabling quantitative analysis of fluorescence intensity, spatial distribution, and morphological parameters.
Applications
Confocal microscopy is widely used in:
Cell and tissue imaging (fixed and live)
Fluorescence colocalization studies
Neuroscience and developmental biology
Immunofluorescence labeling and intracellular tracking
3D reconstruction and quantitative morphometry
Advantages and Limitations
Advantages:
High contrast and resolution via optical sectioning
Reduced background fluorescence
Non-invasive imaging of live specimens
3D and multi-color imaging capability
Quantitative measurement of fluorescence intensity and spatial distribution
Limitations:
Limited excitation wavelengths due to laser availability
High-intensity lasers can damage live cells (mitigated by multiphoton/Nipkow disk systems)
High equipment cost, though entry-level systems and shared core facilities have increased accessibility
Conclusion
Confocal microscopy has revolutionized optical imaging, providing researchers with precise, high-resolution, and multi-dimensional views of biological specimens. By combining advanced lasers, detectors, and computing technology, confocal systems enable insights into cellular and tissue architecture that were previously unattainable, establishing confocal microscopy as an essential tool in modern life sciences.